All in One View
Content from Let's talk about workflows
Last updated on 2025-01-13 | Edit this page
Overview
Questions
- What are the common challenges faced in CMS analysis?
- How can workflow orchestration tools help in capturing the intricate steps involved in producing results in CMS analysis?
Objectives
- Understand the common challenges faced in CMS analysis.
- Learn how workflow orchestration tools can assist in capturing the intricate steps involved in producing results in CMS analysis.
The (short) life of a HEX analysis in CMS
Navigating the complexities of CMS analysis can be a daunting task, regardless of your experience level. A common challenge we all face is capturing the intricate steps involved in producing our results.
Let’s examine the figure below:
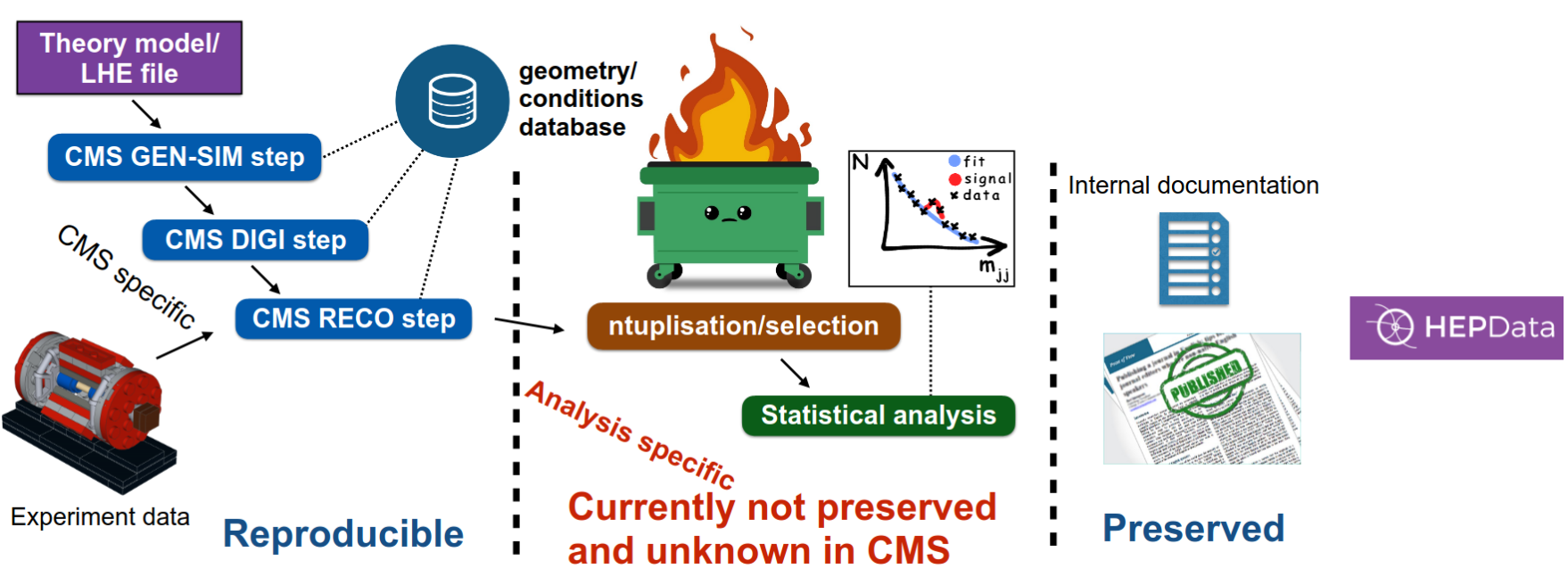
This figure illustrates the typical workflow of a CMS analysis, divided into three distinct phases:
- Collaboration: This encompasses the extensive groundwork laid by the entire research community, over which individual analysts have limited control.
- Analysis: This is the core phase where analysts like ourselves conduct the actual analysis, utilizing the data and resources provided by the collaboration.
- Dissemination: This phase focuses on how our results are shared and utilized by others within the scientific community.
While the dissemination phase is crucial, effective methods for sharing and utilizing research outputs already exist.
In this discussion, I will concentrate on the Analysis phase, which lies entirely within our sphere of influence as analysts. A recurring challenge in high-energy physics (HEX) analysis is the lack of reproducibility. Analysts often embark on a similar (simplified) journey:
- Data Exploration: They begin by analyzing data, generating histograms to visualize distributions.
- Background Modeling: This crucial step involves developing and implementing models to describe background processes.
- Systematic Corrections: Numerous corrections are applied to account for various experimental effects.
- Statistical Analysis: Once the corrected data is obtained, statistical analyses are performed, often utilizing the combine framework.
- Publication: After rigorous peer review, the results are published in scientific journals.
However, this cycle often leads to a significant knowledge gap. When an analyst moves on to a new project or leaves the collaboration, valuable knowledge about the analysis process is lost. Subsequent analysts may struggle to understand (for instance):
- Code Implementation: How the initial histograms were generated and the underlying code.
- Software Dependencies: The specific software versions and configurations used.
- Analysis Workflow: The precise steps involved in applying corrections and performing statistical analyses.
- Tool Usage: How to effectively utilize tools like RooFit or RooStats for the given analysis.
- And several other topics.
This lack of documentation and knowledge transfer hinders efficiency and can lead to unnecessary duplication of effort.
To address some of these reproducibility challenges, the Common Analysis Group (CAT) has developed valuable resources:
- Centralized Code Repository: CAT maintains a central repository for CMS analysis code. This ensures code accessibility and simplifies collaboration among analysts.
- Statistical Interpretation Tools: CAT is working on a suite of tools for preserving and publishing the statistical interpretations of results. These tools promote consistency and transparency in statistical analyses.
By leveraging these resources, analysts can benefit from existing knowledge and code, reducing redundancy and accelerating the analysis process.
Reproducibility
Reproducibility is a cornerstone of scientific research, and HEX is no exception. It ensures that experimental results can be verified independently, bolstering the credibility of scientific findings. To achieve reproducibility, researchers must provide detailed descriptions of their methodologies, including the tools, datasets, and parameters used. This allows others to replicate the experiments and compare their results.
Towards Reusable Analyses: A Three-Step Approach
Let’s explore a three-step approach:
-
Capture Software and Code:
- Objective: Package analysis code and its dependencies for reliable and reproducible execution.
- Current Practices: Many analyses utilize Git (GitHub/GitLab) for code version control. Some leverage containerization technologies (e.g., Docker, Singularity) to encapsulate the entire software environment.
-
Capture Commands:
- Objective: Define the precise commands required to execute the analysis code.
- Current Challenges: Analysis scripts often consist of complex and difficult-to-understand sequences of bash or Python commands.
-
Capture Workflow:
- Objective: Define the ordered execution of commands within the analysis, independent of the specific computing environment.
- Focus: This tutorial will delve into workflow management using modern tools like Snakemake and demonstrate how to execute these workflows on CERN’s REANA platform.
By implementing these steps, we can significantly improve the reproducibility, maintainability, and shareability of our physics analyses.
A quick remark on LAW
While some analysts utilize the Luigi Analysis Workflow LAW, a workflow orchestration tool built on top of Spotify’s Luigi and developed by a CMS physicist, this tutorial focuses on Snakemake. Both LAW and Snakemake are excellent tools, and if you’re already using LAW for your workflow management, that’s great!
What does reproducibility has to do with me?
Who are the first users of a reproducible workflow?
Me, myself and my group. 😁
While we’ve discussed capturing entire CMS analyses, the benefits of reproducible workflows extend beyond best practices.
- Streamlining Repetitive Tasks: Many analyses involve numerous small, repetitive steps. Workflow orchestration tools automate these tasks, saving significant time and resources.
- Facilitating Future Revisions: With increasingly complex analyses and rigorous CMS review processes, the need to revisit and modify earlier steps in the analysis is inevitable. A well-defined workflow ensures that these revisions can be implemented efficiently and accurately, preventing the need to “reinvent the wheel.”
Ultimately, remember that the most frequent user of your analysis code will be your future self.
- Workflow orchestration tools automate repetitive tasks, saving time and resources.
- Well-defined workflows facilitate efficient and accurate revisions in complex analyses.
Content from Snakemake
Last updated on 2025-03-06 | Edit this page
Overview
Questions
- How can I automate complex computational pipelines?
- How can I visualize and understand the structure of my Snakemake workflow?
- How can I create flexible and scalable Snakemake workflows to handle diverse datasets?
Objectives
- To introduce Snakemake as a powerful tool for automating complex computational pipelines.
- To provide a practical guide to creating and executing Snakemake workflows, including the use of rules, wildcards, and configuration files.
- To demonstrate how to visualize and analyze Snakemake workflows
using tools like
--dagand--rulegraphto optimize performance and identify potential bottlenecks.
Snakemake: A Flexible Workflow Engine
Snakemake is a workflow management system that simplifies the creation and execution of complex computational pipelines. It’s particularly useful for bioinformatics pipelines, but can be applied to a wide range of computational tasks. It’s particularly well-suited for tasks involving large datasets and parallel processing, making it a popular choice in fields like high energy physics.
Let’s describe the key features of Snakemake:
- Declarative Syntax: Snakemake uses a declarative language to define workflows, focusing on what you want to achieve rather than how to achieve it. This makes pipelines more readable and maintainable.
- Rule-Based System: Workflows are defined as a series of rules. Each rule represents a task or process, specifying its inputs, outputs, and the command to execute.
- Dependency Management: Snakemake automatically determines the order in which rules need to be executed based on their dependencies. This ensures that tasks are performed in the correct sequence.
- Parallel Execution: Snakemake can efficiently distribute tasks across multiple cores or machines, accelerating the execution of large-scale pipelines.
- Flexibility: It can handle a wide range of computational tasks, from simple data processing to complex simulations.
- Integration with Tools: Snakemake can easily integrate with various tools and libraries used in high energy physics, such as ROOT, TensorFlow, and PyTorch.
Why Snakemake for High Energy Physics?
- Complex Workflows: High energy physics experiments often involve intricate pipelines with numerous steps, from data acquisition and reconstruction to analysis and simulation. Snakemake’s declarative syntax and dependency management make it easy to handle such complex workflows.
- Large Datasets: Snakemake can efficiently process and analyze large datasets generated by high energy physics experiments, thanks to its parallel execution capabilities and integration with data management tools.
- Reproducibility: By defining workflows in a declarative language, Snakemake ensures that results are reproducible. This is crucial in scientific research where experiments need to be verifiable.
- Scalability: Snakemake can scale to handle large-scale computational resources, allowing researchers to efficiently utilize HPC clusters for their analyses.
Understanding the Basics
At its core, Snakemake organizes computational workflows into rules, each representing a specific task within the pipeline. These rules are interconnected through their input and output files, forming a dependency graph. Using a Python-like syntax, you can specify the precise commands required to generate output files from input data. Snakemake intelligently analyzes this dependency graph to determine the optimal execution order and parallelizes tasks to maximize efficiency, making it ideal for large-scale data analysis projects.
The Core Components of a Snakemake Workflow
- Snakefile: This is the main file of a Snakemake workflow. It contains the definition of all rules and config file references. This is where you define your pipeline.
- Config file: This file defines parameters and variables that can be used in your rules. (Useful, but not mandatory.)
- Rules: These define the steps in your pipeline. Each rule has three
important parts:
- Input files: The files that the rule needs to start.
- Output files: The files that the rule will produce.
- Shell command: The command to be executed to produce the output files from the input files.
A Simple Example: A Parallel Workflow
Let’s create a simple workflow that simulates a data analysis pipeline. We’ll have two steps:
- Data Simulation: Simulate some data.
- Data Analysis: Analyze the simulated data.
We’ll use parallel processing to speed up the analysis step.
- Create the Python Scripts:
simulate_data.py:
PYTHON
import sys
sample = sys.argv[1]
with open(f"data/{sample}.txt", "w") as f:
f.write(f"Simulated data for {sample}\n")analyze_data.py:
PYTHON
import sys
with open(sys.argv[1], "r") as f:
data = f.read()
with open(f"results/analysis_{sys.argv[1].split('/')[-1]}", "w") as f:
f.write(f"Analysis of {data}\n")- Create the Config File (config.yaml):
- Create the Snakefile:
YAML
configfile: "config.yaml"
rule all:
input:
expand("results/analysis_{sample}.txt", sample=config["samples"])
rule simulate_data:
output:
"data/{sample}.txt"
shell:
"python simulate_data.py {wildcards.sample} > {output}"
rule analyze_data:
input:
"data/{sample}.txt"
output:
"results/analysis_{sample}.txt"
shell:
"python analyze_data.py {input} > {output}"Explanation of the Workflow:
-
Rules:
- rule simulate_data: This rule simulates data for each sample. As it has no input dependencies, it can be executed at the beginning of the pipeline.
-
rule analyze_data: This rule analyzes the simulated
data for each sample. It depends on the output of the
simulate_datarule, ensuring that this rule only proceeds aftersimulate_datais complete. -
rule all: A rule without an output will
always run, and therefore it is one of the possible
ways to define the final goal of the workflow. (
rule allis the common rule name of the final output) By defining the desired output, Snakemake automatically determines the necessary steps and their execution order. This rule also allows for the use ofwildcards, which enable flexible and scalable workflows.
Parallelism: Snakemake automatically parallelizes the analyze_data rule for each sample, as they are independent of each other.
Wildcards: The
{wildcards.sample}syntax is used to dynamically generate input and output file names based on the sample name.
Running snakemake
Load your environment or container to launch snakemake. Then to run the simple example:
Each part of this command serves a specific purpose:
-
--snakefile: This flag specifies the path to the Snakefile, which contains the definitions of the rules and their dependencies. -
--configfile: This flag indicates the path to the configuration file (YAML format) where you can define parameters and variables that can be used within the Snakefile. (It is not mandatory.) -
--dry-run: This flag tells Snakemake to simulate the workflow execution without actually running the commands. It’s useful for visualizing the execution order of rules and identifying potential issues before running the actual workflow.
OUTPUT
Config file config.yaml is extended by additional config specified via the command line.
Building DAG of jobs...
Job stats:
job count
------------- -------
all 1
analyze_data 5
simulate_data 5
total 11
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample2.txt
jobid: 4
reason: Missing output files: data/sample2.txt
wildcards: sample=sample2
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample3.txt
jobid: 6
reason: Missing output files: data/sample3.txt
wildcards: sample=sample3
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample4.txt
jobid: 8
reason: Missing output files: data/sample4.txt
wildcards: sample=sample4
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample5.txt
jobid: 10
reason: Missing output files: data/sample5.txt
wildcards: sample=sample5
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample1.txt
jobid: 2
reason: Missing output files: data/sample1.txt
wildcards: sample=sample1
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample5.txt
output: results/analysis_sample5.txt
jobid: 9
reason: Missing output files: results/analysis_sample5.txt; Input files updated by another job: data/sample5.txt
wildcards: sample=sample5
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample1.txt
output: results/analysis_sample1.txt
jobid: 1
reason: Missing output files: results/analysis_sample1.txt; Input files updated by another job: data/sample1.txt
wildcards: sample=sample1
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample4.txt
output: results/analysis_sample4.txt
jobid: 7
reason: Missing output files: results/analysis_sample4.txt; Input files updated by another job: data/sample4.txt
wildcards: sample=sample4
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample3.txt
output: results/analysis_sample3.txt
jobid: 5
reason: Missing output files: results/analysis_sample3.txt; Input files updated by another job: data/sample3.txt
wildcards: sample=sample3
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample2.txt
output: results/analysis_sample2.txt
jobid: 3
reason: Missing output files: results/analysis_sample2.txt; Input files updated by another job: data/sample2.txt
wildcards: sample=sample2
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule all:
input: results/analysis_sample1.txt, results/analysis_sample2.txt, results/analysis_sample3.txt, results/analysis_sample4.txt, results/analysis_sample5.txt
jobid: 0
reason: Input files updated by another job: results/analysis_sample3.txt, results/analysis_sample1.txt, results/analysis_sample4.txt, results/analysis_sample2.txt, results/analysis_sample5.txt
resources: tmpdir=<TBD>
Job stats:
job count
------------- -------
all 1
analyze_data 5
simulate_data 5
total 11
Reasons:
(check individual jobs above for details)
input files updated by another job:
all, analyze_data
output files have to be generated:
analyze_data, simulate_data
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.The --dry-run option is a valuable tool for testing your
Snakemake workflow without actually executing the commands. It allows
you to visualize the planned execution order of rules, inspect input and
output files, and verify the use of wildcards and parameters.
To gain even deeper insights into the specific commands that will be
executed, you can employ the --printshellcmds option. This
option will print the shell commands associated with each rule,
providing a detailed breakdown of the actions that Snakemake will
perform.
By combining these options, you can effectively debug, optimize, and fine-tune your Snakemake workflows.
How to validate your workflow
Before diving into the details of your Snakemake workflow, it’s
crucial to validate its design and ensure it functions as expected.
Snakemake offers several tools for this purpose: --dry-run
simulates the workflow, --dag visualizes the dependencies
between rules, and --rulegraph provides a more detailed
view of data flow. By utilizing these tools, you can effectively debug,
optimize, and gain a comprehensive understanding of your pipeline.
--dag
The --dag option generates a Directed Acyclic Graph
(DAG) of your workflow. A DAG is a diagram that illustrates the
dependencies between rules. Each node in the graph represents a rule,
and the edges show the dependencies between rules. By visualizing the
DAG, you can:
- Identify critical paths: Pinpoint the longest sequence of dependent rules that determine the overall workflow duration.
- Detect potential bottlenecks: Identify rules that might limit the overall workflow performance.
- Optimize workflow design: Rearrange rules or adjust parallelism to improve efficiency.
Can you try to visualize this simple pipeline? Try with:
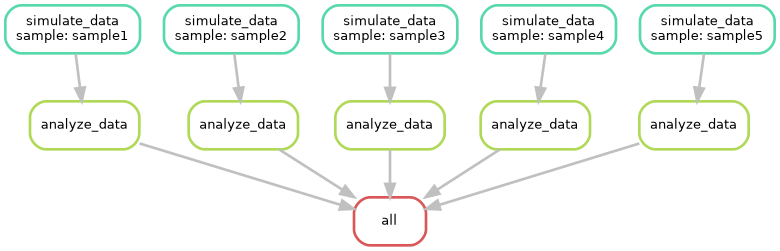
--rulegraph
The --rulegraph option generates a more detailed graph
of your workflow, including information about input and output files.
This can be helpful for understanding the flow of data through your
pipeline.
Can you try to visualize this simple pipeline? Try with:
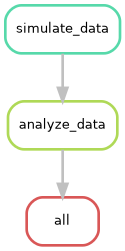
Question:
Can you think about when it can be helpful to use
--dry-run, --dag, or
--rulegraph
Finally, you can run the workflow by removing the
--dry-run flag:
if everything ran succesfully, at the end of the output you will have something like:
OUTPUT
[Thu Nov 14 09:50:51 2024]
Finished job 0.
11 of 11 steps (100%) done
Complete log: .snakemake/log/2024-11-14T095051.589842.snakemake.logIf you got an Snakemake error in the previous command
Depending on the version of Snakemake, you might encounter an error
about the number of cores not being specified. If this happens, add
--cores 1 at the end of the bash command.
You can also notice that you have two folders: data and
results which are the outputs of this simple workflow.
What Happens When You Run Your Workflow Again?
A key feature of Snakemake is its ability to efficiently manage workflow execution based on file timestamps. When you run a Snakemake workflow:
- File Checks: Snakemake first examines the input and output files specified in your rules.
- Dependency Analysis: It then analyzes the dependency graph to determine which rules need to be executed.
- Execution: Only rules whose output files are missing, outdated, or have outdated input files will be executed.
If you rerun the workflow without modifying the input files or deleting the output files, you’ll typically see a message like:
OUTPUT
Nothing to be done (all requested files are present and up to date).This behavior ensures that Snakemake avoids unnecessary computations and efficiently utilizes resources.
More about wildcards
Wildcards are powerful tools in Snakemake that enable you to create flexible and scalable workflows. They act as placeholders within rule names and file paths, allowing you to define generic rules that can handle many different input and output files.
How do wildcards work? You can define wildcards within curly braces {} in your Snakemake file. When Snakemake executes a rule, it replaces the wildcard with specific values, allowing the rule to process multiple files.
Example:
rule analyze_sample:
input:
"data/{sample}.txt"
output:
"results/{sample}_results.txt"
shell:
"python analyze.py {input} {output}"In this example, {sample} is a wildcard. Snakemake will automatically iterate over different sample names and execute the rule for each one, creating specific input and output files.
One can also define wildcards in the rule all rule.
By effectively using wildcards, you can significantly simplify your Snakemake workflows and make them more adaptable to varying datasets and experimental designs.
What’s next?
This is just an overview of the capabilities of Snakemake. As a widely used program, you can find numerous resources online. For more information, you can also visit their official website.
In addition, Snakemake has a public catalog of thousands of workflow in many fields. If you want to see more sophisticated examples, please follow the link to the Snakemake workflow catalog.
- Snakemake automates complex computational pipelines, ensuring efficient resource utilization and avoiding unnecessary computations.
- The Snakefile contains a list of rules describing the pipeline.
- Snakemake manages the workflow execution based on file timestamps.
- Snakemake contains some tools that allows you to debug and validate
your workflows, like
--dry-run,--dag, or--rulegraph.
Content from A simple SUSY analysis
Last updated on 2025-01-13 | Edit this page
Overview
Questions
- Are there more useful Snakemake flags that one can use?
- How does Snakemake ensure a consistent and isolated environment for each rule’s execution when using containerized environments?
Objectives
- Understand the needs of each workflow.
- Understand how to use Snakemake with containerized environments to ensure consistent and isolated execution of workflow rules.
After learning the basics about Snakemake using dummy processes, let’s use a simple example from a SUSY analysis to expand the Snakemake capabilites.
For this part of the tutorial, let’s clone the following repository:
BASH
git clone ssh://git@gitlab.cern.ch:7999/cms-analysis/analysisexamples/snakemake-reana-examples/cmsreana_susyexample.gitIn this example, we’ll demonstrate a basic use case: skimming a nanoAOD file and generating histograms suitable for CMS Combine. While the specific details of this analysis aren’t crucial, this example will highlight REANA’s potential for more complex and sophisticated workflows (more about REANA in the next episode).
A simple SUSY analysis
Let’s discuss the content of the file Snakefile:
output_dir = "."
# Define the final target rule
rule all:
input:
config["output_file"]
# Rule for skimming
rule skimming:
input:
config["nanoaod_file"]
output:
"output/skimming/DY_Skim.root"
params:
selection = '"(nMuon>0&&nTau>0&&HLT_IsoMu24)"',
N = 1000
container:
"docker://gitlab-registry.cern.ch/cms-analysis/analysisexamples/snakemake-reana-examples/cmsreana_susyexample:latest"
shell:
"""
mkdir -p {output_dir}/output/skimming
cd /code/CMSSW_13_0_10/src && \
source /cvmfs/cms.cern.ch/cmsset_default.sh && \
cmsenv && \
python3 PhysicsTools/NanoAODTools/scripts/nano_postproc.py \
{output_dir}/output/skimming {output_dir}/{input} \
--bi /code/CMSSW_13_0_10/src/SUS_ex/Analysis/scripts/keep_in.txt \
--bo /code/CMSSW_13_0_10/src/SUS_ex/Analysis/scripts/keep_out.txt \
-c {params.selection} \
-I SUS_ex.Analysis.DiTau_analysis analysis_mutaumc \
-N {params.N}
"""
# Rule for datacarding
rule datacarding:
input:
"output/skimming/DY_Skim.root"
output:
config['output_file']
params:
year = "2022postEE"
container:
"docker://gitlab-registry.cern.ch/cms-analysis/analysisexamples/snakemake-reana-examples/cmsreana_susyexample:latest"
shell:
"""
mkdir -p {output_dir}/output/datacards
cd /code/CMSSW_13_0_10/src/SUS_ex/Analysis2 && \
source /cvmfs/cms.cern.ch/cmsset_default.sh && \
cmsenv && \
./FinalSelection_mutau.exe {params.year} {output_dir}/{input} {output_dir}/{output} DY DY
"""
The provided example demonstrates a Snakemake workflow with two
rules: skimming and datacarding. One can notice that this workflow
relies on some CMSSW packages, as well as CVMFS environments. While the
specific details of these rules might be analysis-specific, we can focus
on the general concepts of params and
container to enhance workflow flexibility and
reproducibility.
container:
- Encapsulating Environment: By specifying a container image, you create a self-contained environment for your workflow, which can be reproducible. This ensures that the execution environment, including specific software versions and dependencies, is consistent across different systems.
- Simplifying Setup: Using containers eliminates the need for complex installations and configuration on the host system. You can simply pull the container image and run the workflow.
params:
- Parameterizing Rules: The params option allows you to pass parameters to your shell commands, making your rules more flexible and adaptable to different input data or configuration settings.
- Leveraging Configuration Files: By defining parameters in a configuration file, you can easily modify the behavior of your workflow without changing the Snakefile itself.
Containers: The Key to Reproducible Results
Using containerized environments is highly recommended for achieving reproducible research outcomes. Containers offer several advantages:
- Isolation and Consistency: They create a self-contained environment with specific software versions and dependencies, guaranteeing consistent execution across different computing platforms. This eliminates potential issues arising from variations in the host system’s configuration.
- Simplified Setup: Containers eliminate the need for complex installations and environment configuration on the user’s machine. By pulling the pre-built container image, users can readily execute the workflow without worrying about compatibility or missing software.
- Enhanced Sharing: Sharing containerized workflows is straightforward as they encapsulate the entire execution environment. This facilitates collaboration and streamlines research efforts.
Curious about the specific software included in the container used for this analysis? You can delve deeper by examining the Dockerfile located in the repository here: link to Dockerfile.
Let’s validate the workflow:
You can try to run the --dag and/or
--rulegraph commands to visualize what you will run. If it
does like everything is correct, you can try to run it:
Did the workflow finish succesfully?
While we’ve covered the core concepts of Snakemake, there are
numerous additional flags that can be used to customize and optimize
your workflows. One such flag is --use-singularity (or
--use-apptainer in newer Snakemake versions), which is
essential when running rules within containerized environments.
By using --use-apptainer, Snakemake ensures that the
specified container is utilized for each rule’s execution, providing a
consistent and isolated environment for your workflow. Let’s try our
example with
If the previous Snakemake command didn’t execute as expected, it’s likely due to the requirement for access to CERN tools like CVMFS, EOS, or VOMS-proxy. While Snakemake offers some mechanisms to incorporate these tools, REANA provides a more seamless and efficient solution.
In the next episode, we’ll delve deeper into REANA and explore how it simplifies the execution of complex workflows, especially those involving CERN-specific tools and resources.
- Understanding the importance of containerized environments in ensuring consistent and isolated execution of Snakemake workflows.
- Recognizing the role of REANA in simplifying the execution of complex workflows, especially those requiring CERN-specific tools and resources.
Content from Running workflows in REANA
Last updated on 2024-12-30 | Edit this page
Overview
Questions
- What is REANA and how does it enhance the reproducibility of scientific research?
- How does REANA leverage containerization technology and cloud computing resources to simplify the management of computational environments and data dependencies?
Objectives
- Understand how to define, execute, and share complex data analysis workflows using REANA.
- Learn how REANA leverages containerization technology and cloud computing resources to simplify the management of computational environments, data dependencies, and execution pipelines.
REANA is a powerful platform designed to streamline and enhance the reproducibility of scientific research, particularly in high-energy physics analysis. It enables researchers to define, execute, and share complex data analysis workflows, ensuring that their work is transparent, verifiable, and easily replicated. By leveraging containerization technology and cloud computing resources, REANA simplifies the management of computational environments, data dependencies, and execution pipelines. This tutorial will guide you through the essential steps of using REANA, from creating your first workflow to deploying it on a remote computing infrastructure. REANA offers a user-friendly approach to reproducible research, empowering you to focus on scientific discovery while automating the underlying infrastructure.
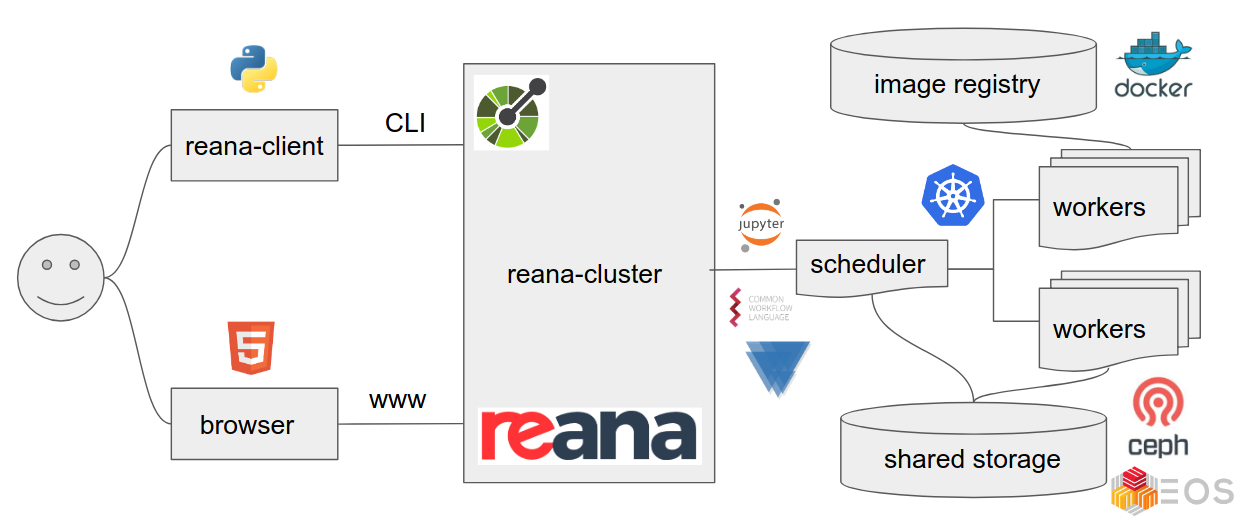
While REANA is primarily a tool for reproducible analysis, it effectively functions as a comprehensive analysis facility, seamlessly integrating various CERN resources. This means that essential CERN tools, including EOS, CVMFS, Kerberos, Rucio, VOMS-proxy, HTCondor, can be directly accessed within the REANA platform, streamlining the analysis process and simplifying resource management.
Understanding the Basics
REANA offers flexibility in workflow management by supporting multiple systems like CWL, Serial, Yadage, and Snakemake. While there’s a growing adoption of Snakemake within the LHC community due to its large external user base and strong support, REANA remains agnostic to the chosen workflow system. However, due to its popularity and powerful features, we will primarily focus on Snakemake throughout this tutorial.
Defining a workflow for REANA
While a Snakemake worflow is defined in a snakefile, in REANA one needs to create a REANA file to include all the parameters that the snakemake workflow will need.
Let’s retake the previous SUSY example, and try to run it using
REANA. First we need to write a reana.yaml file:
YAML
version: 0.9.3
inputs:
directories:
- SUSY/
parameters:
input: inputs.yaml
files:
- Snakefile
workflow:
type: snakemake
file: Snakefile
resources:
cvmfs:
- cms.cern.ch
workspace:
retention_days:
output/*: 30
outputs:
directories:
- output/The reana.yaml file acts as a blueprint for your REANA
workflow, defining essential information for execution.
-
version(optional): Since REANA is under development, specifying the version used (e.g.,0.9.3at the time of writing) can be helpful for troubleshooting. -
inputs: This section defines the files, folders, and parameters your workflow requires. Remember that we need to upload all necessary files before the workflow runs.-
directoriesandfiles: Specify the files and folders to be uploaded to the REANA platform. -
parameters: In this example, workflow parameters are defined in a separateinputs.yamlfile, so we reference the input file here. You can also define parameters directly within reana.yaml.
-
-
workflow: This crucial section tells REANA about the type of workflow you’re using.-
type: we are usingsnakemake, but REANA supports CWL, Serial or Yadage. (More here). -
file: This defines the location of your workflow script. -
resources: Define any global resources required for your workflow execution (detailed information available at here). Remember that you can also define dedicated rule resources in the snakefile.
-
-
workspace(optional): Here, you can set options likeretention_daysto specify how long specific folders should be retained after workflow completion. -
outputs: This section informs REANA which files or folders should be made available for download after successful workflow execution. These can be individual files or entire directories.
More about reana.yaml files can be found
here.
Running a workflow in REANA
Let’s get familiar with the steps necessary to run our workflow in REANA. First, activate the REANA environment, and then remember to set these variables:
export REANA_SERVER_URL=https://reana.cern.ch
export REANA_ACCESS_TOKEN=xxxxxxxxxxxxxxxxxxxxxxxThis needs to be done every time you start a session. Then, the REANA client contains a similar validation than Snakemake’s dry-run, we can run:
OUTPUT
Building DAG of jobs...
[WARNING] Building DAG of jobs...
Job stats:
job count
----------- -------
all 1
datacarding 1
skimming 1
total 3
[WARNING] Job stats:
job count
----------- -------
all 1
datacarding 1
skimming 1
total 3
==> Verifying REANA specification file... /srv/reana.yaml
-> SUCCESS: Valid REANA specification file.
==> Verifying REANA specification parameters...
-> SUCCESS: REANA specification parameters appear valid.
==> Verifying workflow parameters and commands...
-> SUCCESS: Workflow parameters and commands appear valid.
==> Verifying dangerous workflow operations...
-> WARNING: Operation "cd /" found in step "skimming" might be dangerous.
-> WARNING: Operation "cd /" found in step "datacarding" might be dangerous.This step verifies first if the snakefile contains a workflow that
can be run, and second it verifies that the inputs in the
reana.yaml file are correct. If everything looks ok, we can
create a workflow called test_SUSY within the platform:
Remember that this step will only create the workflow within REANA, you can verify it by looking at https://reana.cern.ch/ or by running:
The next step is to upload the files the workflow needs:
and finally we can make it run:
Again, you can check the status of your jobs via the REANA website or
with reana-client status -w test_SUSY.
Unfortunately, not yet
But before throwing your laptop out the window, don’t worry, this failure actually highlights a key feature of REANA. Let’s explore what happened and how to proceed.
More advance REANA commands
In case your workflow did not run succesfully, it is useful to look a
the log files of your jobs. In the reana-client you can
do:
The Easiest Way: The REANA Web Interface
The most user-friendly way to monitor your workflow is through the REANA web interface. Your job’s interface should look similar to this:
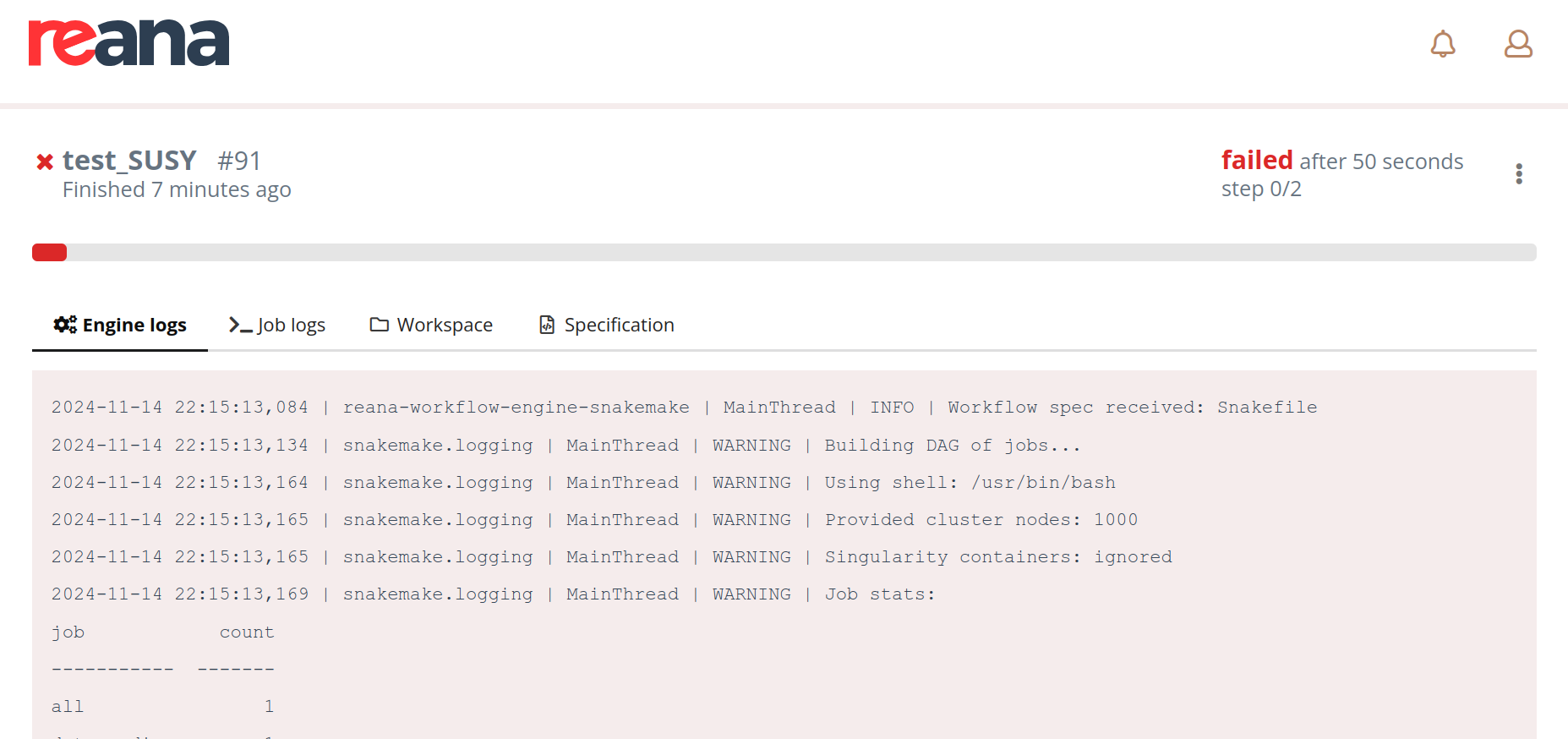
This interface provides valuable insights into your workflow’s status and execution:
- Engine Logs: These logs offer a high-level overview of the workflow’s progress within the REANA Kubernetes cluster. They can be useful for experienced users who want to delve deeper into the technical details.
- Job Logs: Here, you can view the detailed logs for each individual step or rule (in Snakemake terminology) of your workflow. These logs are essential for troubleshooting specific issues.
- Workspace: This section allows you to navigate through the files uploaded to your workflow. It’s a handy tool for debugging and downloading specific files as needed.
- Specification: This provides a high-level overview of your workflow’s configuration in a Python dictionary format. This can be useful for understanding the workflow’s structure and parameters.
Are you getting this error:
OUTPUT
job: :
mkdir: cannot create directory ‘./output’: Permission denied
Error in <TFile::TFile>: file /code/CMSSW_13_0_10/src/./SUSY/DY.root does not existA key feature of REANA-like platforms is that your workflow must be self-contained within the REANA environment. This means that your workflow needs to access its input files and write its output files to specific locations within the REANA workspace.
In this example, notice the following lines at the beginning of the Snakefile:
# Define output folder
# output_dir = "$REANA_WORKSPACE"
output_dir = "."The commented-out line output_dir = "$REANA_WORKSPACE"
demonstrates how to correctly specify the output directory using a REANA
environment variable. REANA provides several environment variables to
help you interact with the platform. A comprehensive list of these
variables can be found in the REANA documentation.
This example demonstrates how to make modifications to your REANA
workflow. In this case, after correcting the output_dir in
your Snakefile, you’ll need to reupload the file to the
REANA platform.
To reupload the modified Snakefile, use
the following command:
You can upload multiple files as needed. Once you’re satisfied with the updated files in your REANA workspace, you can resubmit the workflow:
You can monitor the status of your workflow either through the
command line or the web interface. Note that the workflow name will now
include a number (e.g., test_SUSY.1). This indicates that
you’re running a modified version of the original workflow.
Finally yes!
Checking Workflow Outputs and Reports
Once your workflow completes successfully, you can access its outputs.
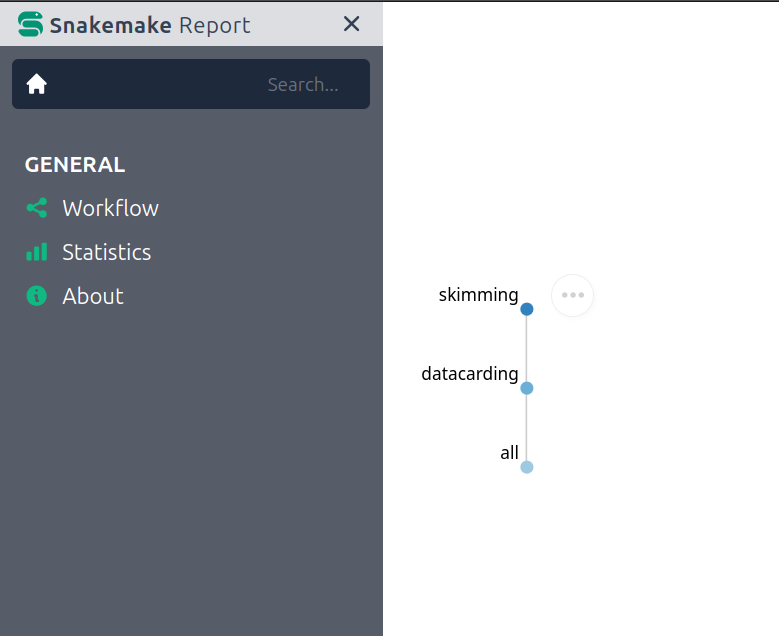
Viewing the Snakemake Report:
A valuable feature of REANA’s Snakemake integration is the generation
of a detailed report. In the REANA web interface, navigate to the
Workspace section and locate the report.html
file. This HTML file provides valuable insights into your workflow’s
execution, including statistics and visualizations.
Downloading Workflow Outputs:
To download the workflow’s output files, you have two options:
- Manual Download:
- Navigate to the Workspace section of the REANA web interface.
- Click on the desired files to download them individually.
- Command-line Download:
- Use the following command to download all output files specified in the reana.yaml file into a compressed ZIP file:
Remember that the download command will only retrieve
the files explicitly listed in the outputs section of your
reana.yaml configuration.
- REANA is more than just a platform for reproducible analysis; it’s a comprehensive analysis facility capable of handling large-scale high-energy physics workflows.
- To ensure reproducibility, it’s crucial to make our workflows independent of the specific analysis facility, such as REANA.
Content from Expanding the SUSY analysis
Last updated on 2024-12-30 | Edit this page
Overview
Questions
- Can we use Snakemake and REANA in more complex examples?
Objectives
- Learn how to expand a SUSY analysis to handle more complex tasks using Snakemake and REANA.
Now that you’ve mastered the basics of creating simple Snakemake workflows and submitting them to REANA, let’s dive deeper into more complex analyses. This next example will illustrate how to expand our SUSY analysis to perform more sophisticated tasks.
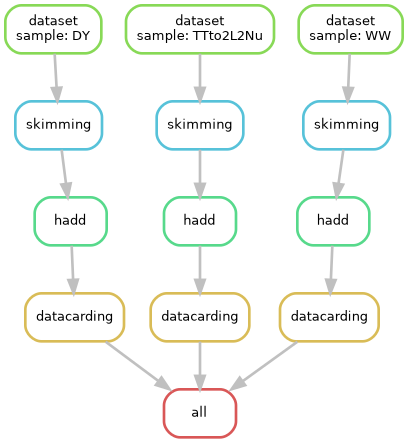
A more real SUSY analysis
In real-world analysis workflows, processing often involves multiple
datasets. Typically, we identify these datasets using CERN’s Data Access
System (DAS) and then extract a list of files for further analysis.
These files are then processed individually before being merged for
subsequent steps. This workflow often involves additional CERN tools
like Rucio for dataset discovery
and manual steps, like using hadd to merge ROOT files.
However, Snakemake and REANA offer a powerful solution to automate and
streamline these tasks. Let’s explore how we can leverage their
capabilities to handle this complex scenario.
Remember
There isn’t a one-size-fits-all approach to creating workflows, just as there isn’t a single way to perform an analysis. The decisions made in this tutorial are primarily for illustrative purposes. While there may be more efficient or optimized ways to structure this workflow, we’ve chosen this approach to clearly demonstrate the core concepts involved.
Let’s look at the Snakefile:
output_dir = "$REANA_WORKSPACE"
analysis_container = "docker://gitlab-registry.cern.ch/cms-analysis/analysisexamples/snakemake-reana-examples/cmsreana_susyexample:latest"
# Define the final target rule
rule all:
input:
expand("output/datacards/{sample}.root", sample=config["samples"])
# Rule for dataset
rule dataset:
output:
"output/dataset_{sample}.txt"
resources:
voms_proxy=True,
rucio=True
params:
sample = "{sample}"
container: "docker://docker.io/reanahub/reana-auth-rucio:1.1.1"
shell:
"""
source datasets.sh {params.sample} ### this is a workaround to get the dataset
mkdir -p {output_dir}/output/
rucio whoami
rucio list-file-replicas cms:$dataset --pfns --protocols root --rses "(tier=2|3)&(country=US|country=DE)" | head -n 5 > {output_dir}/output/dataset_{params.sample}.txt
head {output_dir}/output/dataset_{params.sample}.txt
"""
# Rule for skimming
rule skimming:
input:
"output/dataset_{sample}.txt"
output:
"output/skimming/{sample}/files.txt"
resources:
voms_proxy=True,
kerberos=True,
# compute_backend="htcondorcern"
params:
selection = '"(nMuon>0&&nTau>0&&HLT_IsoMu24)"',
N = 100,
sample = '{sample}'
container: analysis_container
shell:
"""
mkdir -p {output_dir}/output/skimming/{params.sample}
cd /code/CMSSW_13_0_10/src
source /cvmfs/cms.cern.ch/cmsset_default.sh
cmsenv
export X509_CERT_DIR=/cvmfs/grid.cern.ch/etc/grid-security/certificates
echo "Processing files listed in: {input}"
while IFS= read -r file; do
echo "Processing file: $file"
python3 PhysicsTools/NanoAODTools/scripts/nano_postproc.py \
{output_dir}/output/skimming/{params.sample}/ "$file" \
--bi /code/CMSSW_13_0_10/src/SUS_ex/Analysis/scripts/keep_in.txt \
--bo /code/CMSSW_13_0_10/src/SUS_ex/Analysis/scripts/keep_out.txt \
-c {params.selection} \
-I SUS_ex.Analysis.DiTau_analysis analysis_mutaumc \
-N {params.N} --prefetch
done < {output_dir}/{input}
find {output_dir}/output/skimming/{params.sample}/ -type f -name "*root" > {output_dir}/output/skimming/{params.sample}/files.txt
head {output_dir}/output/skimming/{params.sample}/files.txt
"""
# Rule for hadd
rule hadd:
input:
"output/skimming/{sample}/files.txt"
output:
"output/skimming/{sample}_Skim.root"
container: analysis_container
shell:
"""
mkdir -p {output_dir}/output/skimming
cd /code/CMSSW_13_0_10/src && \
source /cvmfs/cms.cern.ch/cmsset_default.sh && \
cmsenv && \
python3 PhysicsTools/NanoAODTools/scripts/haddnano.py \
{output_dir}/output/skimming/{wildcards.sample}_Skim.root \
$(cat {output_dir}/{input})
"""
# Rule for datacarding
rule datacarding:
input:
"output/skimming/{sample}_Skim.root"
output:
"output/datacards/{sample}.root"
params:
year = "2022postEE",
sample = '{sample}'
# sample = lambda wildcards: wildcards.sample
container: analysis_container
shell:
"""
mkdir -p {output_dir}/output/datacards
cd /code/CMSSW_13_0_10/src/SUS_ex/Analysis2 && \
source /cvmfs/cms.cern.ch/cmsset_default.sh && \
cmsenv && \
./FinalSelection_mutau.exe {params.year} {output_dir}/{input} {output_dir}/{output} {params.sample} {params.sample}
"""
Before running this workflow, let’s discuss a few new features introduced in this example:
- Rule-Specific Containers: One of the advantages of containerization is the ability to define specific software environments for each rule. This allows you to use different tools and configurations for different parts of your workflow.
- Consolidated Output Folder: While not strictly necessary, organizing all outputs into a single folder can simplify your workflow and subsequent analysis. We recommend this approach for better organization and easier management.
- Diverse Resource Requirements: To access CMS datasets, Rucio, or other resources, you often need to configure specific credentials and settings. REANA seamlessly integrates with these resources, allowing you to specify the necessary resources for each rule.
REANA resources
Securing Your Credentials with REANA Secrets
To access resources like CMS datasets or Rucio, you’ll need to
provide your credentials, such as VOMS proxies. To ensure the security
of this sensitive information, REANA uses secrets. These
secrets are encrypted and only accessible to your workflow, protecting
your credentials from unauthorized access.
For detailed instructions on how to configure secrets in REANA, please refer to the official REANA documentation:
- VOMS-PROXY: instructions
- kerberos: instructions
- Rucio: instructions
Users can verify which secrets REANA know by running in the terminal:
IMPORTANT INFORMATION ABOUT VOMS-PROXY
The REANA documentation outlines two methods for configuring VOMS-PROXY credentials:
- Automatic Generation: (Recommended approach) Leverages your user certificate and key to automatically generate the VOMS-PROXY when needed. Ensures that your credentials are always up-to-date and valid.
- Manual Upload: Requires you to upload your VOMS-PROXY file to REANA. Less convenient as you’ll need to re-upload the file whenever it expires.
Important: Do not use both methods simultaneously. REANA prioritizes the manually uploaded VOMS-PROXY file, so if it’s expired, you won’t be able to access remote files via XRootD.
We strongly recommend using the automatic generation method for a seamless and secure workflow.
Computer backends
By default, REANA utilizes Kubernetes as its computing backend. This allows you to easily scale your workflows and allocate resources as needed. For instance, to increase the memory allocated to a specific rule, you can specify it in the resources section:
rule skimming:
...
resources:
compute_backend="kubernetes",
kubernetes_memory_limit="8Gi",
...Note that Kubernetes imposes a maximum memory limit of
9.5Gi per container.
For more demanding workloads or specific resource requirements, REANA also supports integration with HTCondor and Slurm. These backends can provide access to larger computing resources and more flexible scheduling options, but their usage is outside the scope of this tutorial. Please refer to the official documentation for more information.
Storage backends
Each REANA user is allocated 300GB of storage space on the platform. (This is visible in under your profile in the web interface). While this is typically sufficient for most workflows, you can leverage REANA’s integration with your personal EOS storage area for larger datasets. To enable this integration, ensure that your Kerberos credentials are configured correctly in your REANA profile.
In this case, the workflow should work perfectly.
More REANA examples
The REANA team has a compilation of examples in their github area. It is well recommended to visit them for more inspiration.
In addition, users can seek for support and help in the following channels:
- REANA official mattemost channel
- REANA forum
- CMS-REANA mattermost channel
- Explore the REANA team’s compilation of examples on their GitHub for more inspiration and advanced use cases.
- Utilize the REANA support channels, such as the official Mattermost channel and forum, for assistance and community support.