Snakemake
Last updated on 2025-03-06 | Edit this page
Overview
Questions
- How can I automate complex computational pipelines?
- How can I visualize and understand the structure of my Snakemake workflow?
- How can I create flexible and scalable Snakemake workflows to handle diverse datasets?
Objectives
- To introduce Snakemake as a powerful tool for automating complex computational pipelines.
- To provide a practical guide to creating and executing Snakemake workflows, including the use of rules, wildcards, and configuration files.
- To demonstrate how to visualize and analyze Snakemake workflows
using tools like
--dagand--rulegraphto optimize performance and identify potential bottlenecks.
Snakemake: A Flexible Workflow Engine
Snakemake is a workflow management system that simplifies the creation and execution of complex computational pipelines. It’s particularly useful for bioinformatics pipelines, but can be applied to a wide range of computational tasks. It’s particularly well-suited for tasks involving large datasets and parallel processing, making it a popular choice in fields like high energy physics.
Let’s describe the key features of Snakemake:
- Declarative Syntax: Snakemake uses a declarative language to define workflows, focusing on what you want to achieve rather than how to achieve it. This makes pipelines more readable and maintainable.
- Rule-Based System: Workflows are defined as a series of rules. Each rule represents a task or process, specifying its inputs, outputs, and the command to execute.
- Dependency Management: Snakemake automatically determines the order in which rules need to be executed based on their dependencies. This ensures that tasks are performed in the correct sequence.
- Parallel Execution: Snakemake can efficiently distribute tasks across multiple cores or machines, accelerating the execution of large-scale pipelines.
- Flexibility: It can handle a wide range of computational tasks, from simple data processing to complex simulations.
- Integration with Tools: Snakemake can easily integrate with various tools and libraries used in high energy physics, such as ROOT, TensorFlow, and PyTorch.
Why Snakemake for High Energy Physics?
- Complex Workflows: High energy physics experiments often involve intricate pipelines with numerous steps, from data acquisition and reconstruction to analysis and simulation. Snakemake’s declarative syntax and dependency management make it easy to handle such complex workflows.
- Large Datasets: Snakemake can efficiently process and analyze large datasets generated by high energy physics experiments, thanks to its parallel execution capabilities and integration with data management tools.
- Reproducibility: By defining workflows in a declarative language, Snakemake ensures that results are reproducible. This is crucial in scientific research where experiments need to be verifiable.
- Scalability: Snakemake can scale to handle large-scale computational resources, allowing researchers to efficiently utilize HPC clusters for their analyses.
Understanding the Basics
At its core, Snakemake organizes computational workflows into rules, each representing a specific task within the pipeline. These rules are interconnected through their input and output files, forming a dependency graph. Using a Python-like syntax, you can specify the precise commands required to generate output files from input data. Snakemake intelligently analyzes this dependency graph to determine the optimal execution order and parallelizes tasks to maximize efficiency, making it ideal for large-scale data analysis projects.
The Core Components of a Snakemake Workflow
- Snakefile: This is the main file of a Snakemake workflow. It contains the definition of all rules and config file references. This is where you define your pipeline.
- Config file: This file defines parameters and variables that can be used in your rules. (Useful, but not mandatory.)
- Rules: These define the steps in your pipeline. Each rule has three
important parts:
- Input files: The files that the rule needs to start.
- Output files: The files that the rule will produce.
- Shell command: The command to be executed to produce the output files from the input files.
A Simple Example: A Parallel Workflow
Let’s create a simple workflow that simulates a data analysis pipeline. We’ll have two steps:
- Data Simulation: Simulate some data.
- Data Analysis: Analyze the simulated data.
We’ll use parallel processing to speed up the analysis step.
- Create the Python Scripts:
simulate_data.py:
PYTHON
import sys
sample = sys.argv[1]
with open(f"data/{sample}.txt", "w") as f:
f.write(f"Simulated data for {sample}\n")analyze_data.py:
PYTHON
import sys
with open(sys.argv[1], "r") as f:
data = f.read()
with open(f"results/analysis_{sys.argv[1].split('/')[-1]}", "w") as f:
f.write(f"Analysis of {data}\n")- Create the Config File (config.yaml):
- Create the Snakefile:
YAML
configfile: "config.yaml"
rule all:
input:
expand("results/analysis_{sample}.txt", sample=config["samples"])
rule simulate_data:
output:
"data/{sample}.txt"
shell:
"python simulate_data.py {wildcards.sample} > {output}"
rule analyze_data:
input:
"data/{sample}.txt"
output:
"results/analysis_{sample}.txt"
shell:
"python analyze_data.py {input} > {output}"Explanation of the Workflow:
-
Rules:
- rule simulate_data: This rule simulates data for each sample. As it has no input dependencies, it can be executed at the beginning of the pipeline.
-
rule analyze_data: This rule analyzes the simulated
data for each sample. It depends on the output of the
simulate_datarule, ensuring that this rule only proceeds aftersimulate_datais complete. -
rule all: A rule without an output will
always run, and therefore it is one of the possible
ways to define the final goal of the workflow. (
rule allis the common rule name of the final output) By defining the desired output, Snakemake automatically determines the necessary steps and their execution order. This rule also allows for the use ofwildcards, which enable flexible and scalable workflows.
Parallelism: Snakemake automatically parallelizes the analyze_data rule for each sample, as they are independent of each other.
Wildcards: The
{wildcards.sample}syntax is used to dynamically generate input and output file names based on the sample name.
Running snakemake
Load your environment or container to launch snakemake. Then to run the simple example:
Each part of this command serves a specific purpose:
-
--snakefile: This flag specifies the path to the Snakefile, which contains the definitions of the rules and their dependencies. -
--configfile: This flag indicates the path to the configuration file (YAML format) where you can define parameters and variables that can be used within the Snakefile. (It is not mandatory.) -
--dry-run: This flag tells Snakemake to simulate the workflow execution without actually running the commands. It’s useful for visualizing the execution order of rules and identifying potential issues before running the actual workflow.
OUTPUT
Config file config.yaml is extended by additional config specified via the command line.
Building DAG of jobs...
Job stats:
job count
------------- -------
all 1
analyze_data 5
simulate_data 5
total 11
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample2.txt
jobid: 4
reason: Missing output files: data/sample2.txt
wildcards: sample=sample2
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample3.txt
jobid: 6
reason: Missing output files: data/sample3.txt
wildcards: sample=sample3
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample4.txt
jobid: 8
reason: Missing output files: data/sample4.txt
wildcards: sample=sample4
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample5.txt
jobid: 10
reason: Missing output files: data/sample5.txt
wildcards: sample=sample5
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule simulate_data:
output: data/sample1.txt
jobid: 2
reason: Missing output files: data/sample1.txt
wildcards: sample=sample1
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample5.txt
output: results/analysis_sample5.txt
jobid: 9
reason: Missing output files: results/analysis_sample5.txt; Input files updated by another job: data/sample5.txt
wildcards: sample=sample5
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample1.txt
output: results/analysis_sample1.txt
jobid: 1
reason: Missing output files: results/analysis_sample1.txt; Input files updated by another job: data/sample1.txt
wildcards: sample=sample1
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample4.txt
output: results/analysis_sample4.txt
jobid: 7
reason: Missing output files: results/analysis_sample4.txt; Input files updated by another job: data/sample4.txt
wildcards: sample=sample4
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample3.txt
output: results/analysis_sample3.txt
jobid: 5
reason: Missing output files: results/analysis_sample3.txt; Input files updated by another job: data/sample3.txt
wildcards: sample=sample3
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule analyze_data:
input: data/sample2.txt
output: results/analysis_sample2.txt
jobid: 3
reason: Missing output files: results/analysis_sample2.txt; Input files updated by another job: data/sample2.txt
wildcards: sample=sample2
resources: tmpdir=<TBD>
[Wed Nov 13 15:19:33 2024]
rule all:
input: results/analysis_sample1.txt, results/analysis_sample2.txt, results/analysis_sample3.txt, results/analysis_sample4.txt, results/analysis_sample5.txt
jobid: 0
reason: Input files updated by another job: results/analysis_sample3.txt, results/analysis_sample1.txt, results/analysis_sample4.txt, results/analysis_sample2.txt, results/analysis_sample5.txt
resources: tmpdir=<TBD>
Job stats:
job count
------------- -------
all 1
analyze_data 5
simulate_data 5
total 11
Reasons:
(check individual jobs above for details)
input files updated by another job:
all, analyze_data
output files have to be generated:
analyze_data, simulate_data
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.The --dry-run option is a valuable tool for testing your
Snakemake workflow without actually executing the commands. It allows
you to visualize the planned execution order of rules, inspect input and
output files, and verify the use of wildcards and parameters.
To gain even deeper insights into the specific commands that will be
executed, you can employ the --printshellcmds option. This
option will print the shell commands associated with each rule,
providing a detailed breakdown of the actions that Snakemake will
perform.
By combining these options, you can effectively debug, optimize, and fine-tune your Snakemake workflows.
How to validate your workflow
Before diving into the details of your Snakemake workflow, it’s
crucial to validate its design and ensure it functions as expected.
Snakemake offers several tools for this purpose: --dry-run
simulates the workflow, --dag visualizes the dependencies
between rules, and --rulegraph provides a more detailed
view of data flow. By utilizing these tools, you can effectively debug,
optimize, and gain a comprehensive understanding of your pipeline.
--dag
The --dag option generates a Directed Acyclic Graph
(DAG) of your workflow. A DAG is a diagram that illustrates the
dependencies between rules. Each node in the graph represents a rule,
and the edges show the dependencies between rules. By visualizing the
DAG, you can:
- Identify critical paths: Pinpoint the longest sequence of dependent rules that determine the overall workflow duration.
- Detect potential bottlenecks: Identify rules that might limit the overall workflow performance.
- Optimize workflow design: Rearrange rules or adjust parallelism to improve efficiency.
Can you try to visualize this simple pipeline? Try with:
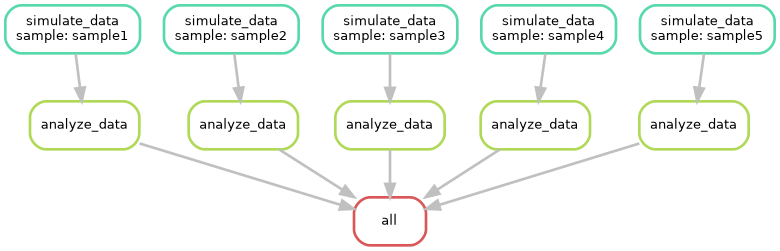
--rulegraph
The --rulegraph option generates a more detailed graph
of your workflow, including information about input and output files.
This can be helpful for understanding the flow of data through your
pipeline.
Can you try to visualize this simple pipeline? Try with:
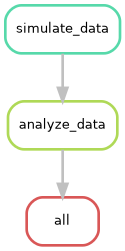
Question:
Can you think about when it can be helpful to use
--dry-run, --dag, or
--rulegraph
Finally, you can run the workflow by removing the
--dry-run flag:
if everything ran succesfully, at the end of the output you will have something like:
OUTPUT
[Thu Nov 14 09:50:51 2024]
Finished job 0.
11 of 11 steps (100%) done
Complete log: .snakemake/log/2024-11-14T095051.589842.snakemake.logIf you got an Snakemake error in the previous command
Depending on the version of Snakemake, you might encounter an error
about the number of cores not being specified. If this happens, add
--cores 1 at the end of the bash command.
You can also notice that you have two folders: data and
results which are the outputs of this simple workflow.
What Happens When You Run Your Workflow Again?
A key feature of Snakemake is its ability to efficiently manage workflow execution based on file timestamps. When you run a Snakemake workflow:
- File Checks: Snakemake first examines the input and output files specified in your rules.
- Dependency Analysis: It then analyzes the dependency graph to determine which rules need to be executed.
- Execution: Only rules whose output files are missing, outdated, or have outdated input files will be executed.
If you rerun the workflow without modifying the input files or deleting the output files, you’ll typically see a message like:
OUTPUT
Nothing to be done (all requested files are present and up to date).This behavior ensures that Snakemake avoids unnecessary computations and efficiently utilizes resources.
More about wildcards
Wildcards are powerful tools in Snakemake that enable you to create flexible and scalable workflows. They act as placeholders within rule names and file paths, allowing you to define generic rules that can handle many different input and output files.
How do wildcards work? You can define wildcards within curly braces {} in your Snakemake file. When Snakemake executes a rule, it replaces the wildcard with specific values, allowing the rule to process multiple files.
Example:
rule analyze_sample:
input:
"data/{sample}.txt"
output:
"results/{sample}_results.txt"
shell:
"python analyze.py {input} {output}"In this example, {sample} is a wildcard. Snakemake will automatically iterate over different sample names and execute the rule for each one, creating specific input and output files.
One can also define wildcards in the rule all rule.
By effectively using wildcards, you can significantly simplify your Snakemake workflows and make them more adaptable to varying datasets and experimental designs.
What’s next?
This is just an overview of the capabilities of Snakemake. As a widely used program, you can find numerous resources online. For more information, you can also visit their official website.
In addition, Snakemake has a public catalog of thousands of workflow in many fields. If you want to see more sophisticated examples, please follow the link to the Snakemake workflow catalog.
- Snakemake automates complex computational pipelines, ensuring efficient resource utilization and avoiding unnecessary computations.
- The Snakefile contains a list of rules describing the pipeline.
- Snakemake manages the workflow execution based on file timestamps.
- Snakemake contains some tools that allows you to debug and validate
your workflows, like
--dry-run,--dag, or--rulegraph.